Sequence Text Search using Codondex
At Codondex one of our central metrics that we have developed is known as iScore. Please see iScore Documentation to view how we calculate iScore.
Like a kmer, but variable we start from (L)ength 7. Assuming a sequence L=400 we compute 399 398, 397...7 kmers. Then, for every kmer we query the kmer subsequence in all kmers and count recurrence.

The red text in the image above represents the variable length kmer, analyzed per offset/window.
The iScore algorithm will count all of these kmer’s as well as their recurrence in other kmers generated from other parts of the base sequence.
We also use a metric called the theoretical iScore which are the recurrence due to the patterning algorithm. In this post we are not going to go into the mechanics of how this is calculated. We can then subtract the real iScore from this theoretical iScore to find how many recurrences from other parts of the base transcript.
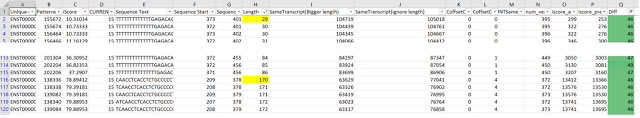
Using this system we can then isolate parts of the sequence with highest recurrence in other parts of the base transcript and select and filter by length of the subsequence (kmer) and start and end positions in the base sequence.
Here a sample table which was developed using these metrics with some filtering.
No comments:
Post a Comment