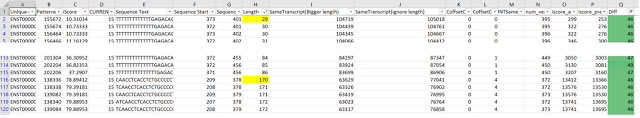
16,904,172 theoretical k-mers were computed for 12 transcripts of Tp53 downloaded from ensembl. Each transcripts' k-mer set contains 1,408,681 entries computed in the same nucleotide order. k-mers were associated with a signature of the transcript's protein. The protein signatures were computed using the same process.
As previously described a ranking based on recurrence was computed for each k-mer using iScore algorithms. We used a vector which we call PiV to assign each k-mer to a position in the order of the 12 k-mers each for a single transcript.
The chart represents, for each transcript the distribution of its k-mers to positions 1-12 based on the iScore and protein signatures. For the 1,408,681 k-mers, at least one position in the vector is dominant in each transcript.