In the chart below, transcript k-mers that demonstrated sequential position in iScore vector (PiV) stability produced uninterrupted lines. k-mer stability/instability is a function of iScore (discussed above) and plot of the sequence of k-mer PiV [value's].
We plotted each k-mer PiV for each transcript. For each transcript, each k-mer can occupy one of twelve positions and each PiV for all transcripts must be occupied 2765 times (see last chart below)

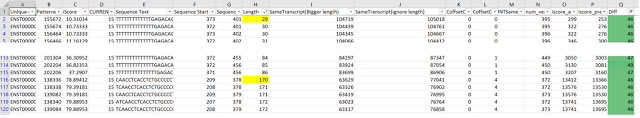
At row 2459 above (top left), there is a single line perturbation (not visible in this image). We traced it to k-mer (#939134) where we discovered that the addition of "G" at nucleotide 1377 caused Transcript T793 to change order with T597, T465. The result of "G" was an iSocre change that caused the order of transcripts to flip.

The next image demonstrate the total PiV (row1) distributions for all 2765 k-mers of each transcript (col N).