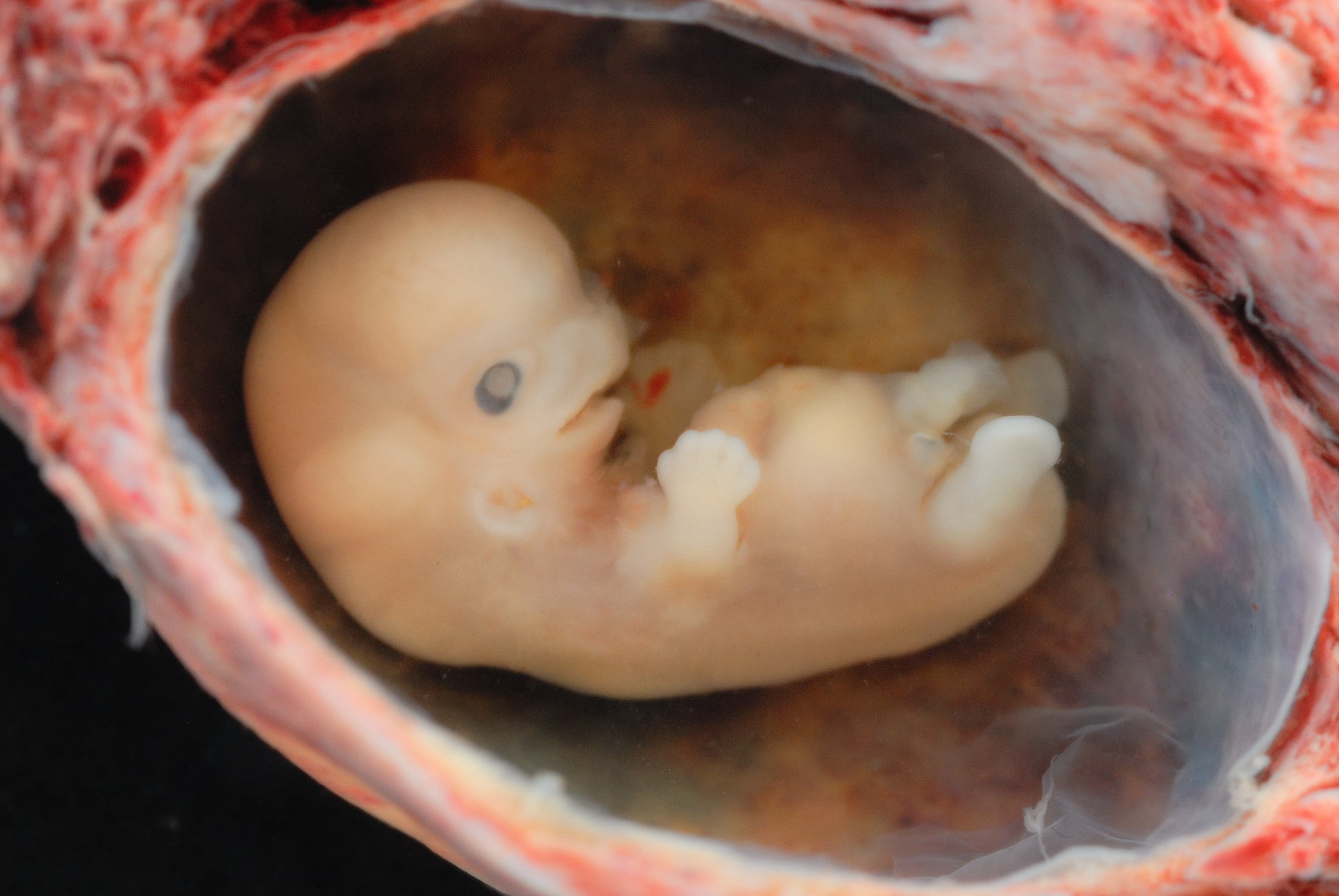
A p53 orchestrated mechanism is required for a blastocyst to implant and penetrate the uterine wall. A trophoblast induced, uterine Natural Killer cell (NK) response against endothelial cells follows resulting in significant vascular remodeling and immune suppression. Similarities parallel p53 in cancer stem cells that influence fibroblast pro-invasiveness, recruit NK to invade endothelial cells during angiogenesis, vascular remodeling and immune suppression. The previous blog entry and various papers follow in support.
Leukemia inhibitory factor (LIF) and LIF receptor expression in human endometrium suggests an autocrine/paracrine function in regulating embryo implantation. The necessary expression of LIF, under p53 control peaks coincidental to blastocyst implantation inviting the question whether blastocyst invokes a paracrine response in endothelial cells of the endometrium?
In mice p53 plays an important role in maternal reproduction through transcriptional regulation of LIF, a cytokine required for blastocyst implantation. To determine whether observations could be extended to humans, a list of single-nucleotide polymorphisms (SNPs) in the p53 pathway that could modify the function of p53 was assembled and used to study their impact on human fertility. Indeed, there is evidence for p53 in reproduction and fecundity. Recent studies with haplotypes of SNP’s in the Mdm4 and Hausp genes also demonstrated the positive evolutionary selection toward alleles in Caucasian populations. These observations suggested p53 has evolutionary conserved functions. p53-like transcription factors are conserved from invertebrates to vertebrates, and the existence of p53-like proteins in short-lived organisms that do not exhibit adult cancer, such as flies and worms suggests that tumor suppression was not the original function for p53 and its pathway.
If SNPs in the p53 pathway regulate human fertility under selective pressure it may suggest similar for immunity and cancer where p53 is the most mutated of all gene’s. The identification of functional SNPs that mediate the p53 stress response is challenging, as there are more than 50,000 SNPs in the NCBI SNP repository (dbSNP) in genes that have been implicated in mediating and regulating the p53 response (Vazquez et al. 2008)
LIF was also identified as a tumor promoter that mediates pro-invasive activation of stromal fibroblasts. It was demonstrated that a pulse of transforming growth factor beta (TGF-β) established stable pro-invasive fibroblast activation by inducing LIF production in both fibroblasts and tumor cells. LIF, a member of the IL-6 proinflammatory cytokine family is the main driver of proinvasive TGF-β-dependent evolution of the tumor microenvironment. LIF mediates autocrine TGF-β1-dependent pro-invasive activation in fibroblasts, whereas, in a paracrine manner, tumor-secreted LIF promotes and sustains pro-invasive conversion of fibroblast.
The proinflammatory cytokine LIF reprograms fibroblasts into a pro-invasive phenotype, which promotes extracellular matrix remodelling and collective invasion of cancer cells. Recently Adorno et al. demonstrated that the mutational status of p53 determines the nature of the cellular response to TGF-β. Introduction of wild-type p53 into p53 null H1299 cells resulted in a TGF-β-induced growth arrest via p21. In contrast reconstitution with mutant p53 caused cells to change from an epithelial to mesenchymal morphology, enabling a promigratory TGF-β response, TGF-β is expressed both in endometrial and trophoblastic cells of the blastocyst. TGF-β was shown to inhibit trophoblast proliferation and invasion apparently by stimulating TIMP secretion and decreasing MMP activation through downregulation of plasminogen activators. In another study TGF-β was found to inhibit trophoblast invasion by reducing MMP-9 and uPA secretion, but did not affect TIMP levels or cell proliferation. Elevated TGF-β activity has been reported in the plasma of pre-eclamptic mothers and may be implicated in the impaired implantation associated with pre-eclampsia.
Anergized NK through secreted cytokine factors and direct cell to cell contact have the ability to induce differentiation of stem cells including squamous cancer stem cells resulting in resistance or vulnerability to NK mediated cytotoxicity.
Peptides derived from p53 are presented by class I MHC molecules and may act as tumor-associated epitopes which could be targeted by p53-specific T cells. Differential T cell recognition patterns of p53 proteins, measured by IFN𝛾 secretion seems to be confined to p53 as an antigen as expression of different p53 mutants neither altered HLA-A0201 surface expression nor impacted on the recognition of another (control) antigen (MART-1). Our results show that selected p53 mutations altering protein stability can modulate p53 presentation to T cells, leading to a differential immune reactivity inversely correlated with measured p53 protein levels. Thus, p53 may behave differently than other classical tumor antigens and its mutational status should therefore be taken into account when elaborating immunotherapy treatments of cancer patients targeting p53.
These paracrine and autocrine interactions describe some of p53’s unique, highly specific signaling capabilities in immune and reproductive control and reorganization. In both, host immune cells are recruited by cytokines that are expressed by invading trophoblasts or cancer stem cells. These invading cells modify, or kill host endothelial cells resulting in a p53 orchestrated phenotype that permits invading cells to remodel capillaries to manage immune responses that would otherwise destabilize the tumor microenvironment or endometrium.
The signaling capabilities of p53 are the target of Precision Autology's Codondex algorithms for use in cell selection and therapy.
No comments:
Post a Comment