Never in the field of molecular oncology have so many sites of posttranslational modification in one protein (p53) been modified by so many different enzymes, but direct response mechanisms that increase immune receptors are rarely discovered and have important implications.
In the tumor microenvironment (TME), cancer associated fibroblasts (CAFs) display an activated phenotype and can physically remodel the extracellular matrix (ECM). Silencing p53 in the CAFs strongly compromised this activity, implicating p53 as a key contributor to a distinctive CAF feature. Here, the non-autonomous, tumor-suppressive activity of non-mutant p53 cDNA is rewired to become a significant contributor to the CAFs’ tumor-supportive activities. This surprising role for p53 in CAFs suggests that, during tumor progression p53 functionality is altered, not only in the cancer cells, but also in their adjacent stroma.
Although p53 is not mutated in the human placenta, it has become functionally incompetent. Why and how p53 is functionally incompetent in cytotrophoblast cells might well be the key to understanding trophoblast invasion. Vascular remodeling for placentation is controlled by small populations of conventional Natural Killer cells, distinct from much larger populations of uterine NK cells, that acidify the ECM with a2V-ATPase, that activates MMP9, degrades the ECM and releases stored pro-angiogenesis growth factors. Similarly hypoxic TME's that in NK cells sustain excessive mitochondrial fission resulting in fragmentation could cause a2V-ATP activated MMP9 to similarly degrade ECM and promote angiogenesis in the early TME.
Another MMP protein, MMP2 is a ligand for the Toll-like receptor 2 (Tlr2). Expression of Tlr2 and Tlr4 in the TME is important for the promotion of tumor growth, and when both of these receptors are absent, growth is compromised. Furthermore, the expression of Tlr2 and Tlr4 in both hematopoietic and stromal compartments appears to support MMP2-driven tumor growth.
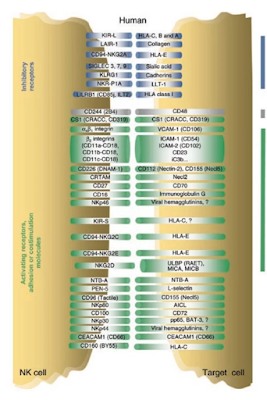
The integration of the TLR gene family into the p53 regulatory network is unique to primates. p53 promoter response elements that are targeted by this DNA damage and stress-responsive regulator suggest a general p53 role in the control of human TLR gene expression. TLR genes show responses to DNA damage, and most are p53-mediated. TLR's mediate innate immunity to a wide variety of threats through recognition of conserved pathogen-associated molecular motifs. Expression of all TLR genes, in blood lymphocytes and alveolar macrophages from healthy volunteers can be induced by DNA metabolic stressors with considerable inter-individual variability. Most TLR genes respond to p53 via canonical as well as noncanonical promoter binding sites.
A polymorphism in a TLR8 response element provided the first human example of a p53 target sequence specifically responsible for endogenous gene induction. These findings—demonstrating that the human innate immune system, including downstream induction of cytokines, can be modulated by DNA metabolic stress—have many implications for health and disease, as well as for understanding the evolution of DNA damage and p53 responsive networks. That p53 can directly increase an inflammatory response differs from the generally held view relating to the antagonistic affect of p53 on inflammation directed by NF-κB. However, the direct mechanism here is different in that it involves another p53-mediated increase in a receptor that translates ligand interactions into cytokine responses.