One of the more intriguing patterns in biology is that processes for normal development often resemble those that appear in disease. Few examples illustrate this better than the similarity between trophoblast invasion during pregnancy and the early stages of tumor growth.
In both situations, cells penetrate surrounding tissue, remodel blood vessels, and establish themselves within an environment that tolerates their presence rather than destroying them. The mechanisms that allow this to occur remain incompletely understood. Increasing evidence suggests that deubiquitinases (DUBs), enzymes that remove ubiquitin from proteins and thereby regulate signaling thresholds, may play an important role in stabilizing this permissive state.
This raises a provocative possibility: the regulatory machinery that enables the maternal–fetal interface to tolerate trophoblast invasion may share features with the mechanisms tumors exploit to evade immune detection.
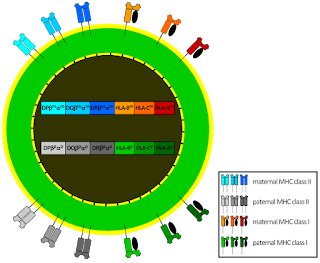
During early pregnancy, decidual natural killer cells (dNK) become the dominant immune cell population in the uterus. Rather than behaving as cytotoxic killers, these cells adopt a distinct phenotype that supports; angiogenesis, spiral artery remodeling, trophoblast invasion.
The density of NK cells in the decidua is striking, often representing 50–70% of immune cells in early pregnancy. Instead of attacking invading trophoblasts, these NK cells participate in building the placenta and converting maternal spiral arteries into vessels capable of supporting fetal circulation.
Maintaining such a high density of NK cells without triggering immune destruction requires a carefully tuned balance between activation signals and inhibitory regulatory pathways. One of the central signals controlling NK cells in both peripheral tissues and the uterus is IL-15. In the decidua, IL-15 produced by stromal cells supports the recruitment, proliferation and survival of NK cells.
Recent work has identified YTHDF2, an m⁶A RNA-binding protein, as a key downstream regulator of this process. In NK cells: IL-15 → STAT5 → YTHDF2 → NK-cell homeostasis. YTHDF2 regulates the stability of specific mRNAs that determine NK survival, proliferation and maturation. Through selective RNA decay, YTHDF2 effectively tunes the functional state of NK cells.
A p53 regulatory layer likely intersects with this system. p53 is best known as a tumor suppressor that responds to DNA damage and cellular stress by regulating transcriptional programs controlling cell cycle arrest and apoptosis. But, p53 also plays an important role in immune signaling and communication between stressed cells and the immune system.
For example, p53 activation can influence immune surveillance by inducing chemokines and inflammatory mediators that recruit immune cells, including NK cells. This places p53 upstream of many of the stress-response pathways that determine whether an NK cell should eliminate a target.
While RNA regulation shapes the NK-cell transcriptome, a second regulatory layer operates through ubiquitin signaling. Many proteins involved in immune activation are controlled by ubiquitination. Deubiquitinases (DUBs) reverse this process, stabilizing proteins or suppressing signaling cascades depending on the target. One DUB that has recently drawn attention is USP13 that has been shown to regulate several pathways central to immune signaling and cellular stress responses, including STING-dependent innate immune activation. Network analysis in prostate cancer datasets also show a strong interaction between USP13 and the RNA regulator YTHDF2, linking ubiquitin signaling to the RNA regulatory machinery governing NK cells.
Interestingly, the relationship between NK cells and invasive cells is not unique to pregnancy. Studies show that NK cells often accumulate in tissues surrounding early tumors, particularly during the earliest stages of transformation. In many cancers, NK cells are present in peritumoral tissue, but become functionally suppressed or excluded as tumors progress. This pattern suggests that the immune system initially recognizes abnormal cells but may later be restrained by tumor-driven immunoregulatory mechanisms. The result is a paradox: NK cells are present but ineffective.
Taken together, these observations suggest a regulatory architecture that could stabilize environments where invasion must occur without triggering destructive immunity.
In such a system:
Cellular stress signals activate p53 and generate stress-response transcripts.
Endogenous repeat RNAs may activate innate immune sensing pathways such as RIG-I, MDA5 and STING.
Cytokine signaling such as IL-15 supports NK-cell expansion and survival.
RNA-level regulation via YTHDF2 tunes NK-cell gene expression and maturation.
Deubiquitinases such as USP13 modulate innate immune signaling intensity and prevent excessive inflammatory activation.
The combined effect could be a high-NK-density but low-cytotoxic environment capable of supporting tissue remodeling and vascular development. In pregnancy, this environment enables trophoblast cells to invade maternal tissue and establish the placenta. Tumors may exploit the same architecture
Early tumors face a challenge similar to that encountered by trophoblasts: they must expand and invade tissue while avoiding immune elimination.
Many tumors exhibit features reminiscent of the decidual microenvironment, including; suppressed innate immune signaling, dysfunctional or tolerized NK cells, enhanced angiogenesis and extensive tissue remodeling. If DUBs such as USP13 help establish these permissive states, tumors could potentially co-opt the same regulatory circuits that operate at the maternal–fetal interface.
In this view, tumors may hijack a developmental program that normally allows pregnancy to proceed successfully.
The decidua represents one of the most extreme natural examples of immune tolerance in mammals. Understanding how this system maintains large NK-cell populations without triggering inflammation could reveal new strategies for controlling immune responses in other contexts.
If deubiquitinase signaling and p53-mediated nucleic-acid sensing help stabilize this balance, they may represent a broader biological principle; the same regulatory networks that enable successful pregnancy may also be exploited by tumors to evade immune detection.
Recent studies showing that rye-derived alkylresorcinols activate SIRT3-mediated autophagy and restore mitochondrial function suggest that metabolic stress regulation may sit upstream of the inflammatory and immune circuits that govern both trophoblast implantation and tumor invasion.
Uncovering these shared mechanisms could deepen our understanding of both reproductive biology and cancer immunology, and potentially reveal new therapeutic strategies in the process.