 |
| Immune cells get comfortable with cancer Courtesy https://deepai.org |
A hallmark of cancer, autoimmunity and disease is the aberrant transcription of typically silenced, repetitive genetic elements that mimic Pathogen-Associated Molecular Patterns (PAMP's) that bind Pattern Recognition Receptors (PPR's) triggering the innate immune system and inflammation. Unrestrained, this 'viral mimicry' activates a generally conserved mechanism that, under restraint, supports homeostasis. These repetitive viral DNA sequences normally act as a quality control over genomic dysregulation responding in ways that preferentially promote immune conditions for stability. If aberrantly unrestrained and the 'viral mimicry' is transcribed it may result in undesirable immune reactions that disrupt the homeostasis of cells.
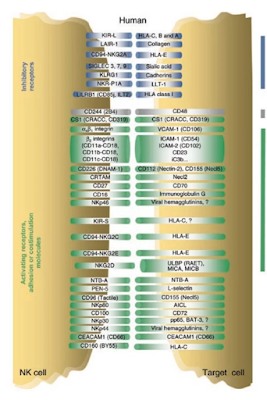
Mitochondrial DNA (mtDNA) are one source of cytosolic double stranded RNA (dsRNA) that is commonly present in cells. Trp53 Mutant Embryonic Fibroblasts (MEF's) contain innate immune stimulating endogenous dsRNA, from mtDNA that mimic PAMP's. The immune response, via RIG-1 like PRR, leads to expression of type 1 interferon (IFN) and proinflammatory cytokine genes. Further, Natural Killer cells also produce a multitude of cytokines that can promote or dampen an immune response. Wild-type p53 suppresses viral repeats and contributes to innate immunity by enhancing IFN-dependent antiviral activity independent of its function as a proapoptotic and tumor suppressor gene.
Post-translationally modified P53, located in the cytoplasm, enhances the permeability of the mitochondrial outer membrane thus stimulating apoptosis. However, treating Trp53 mutant MEF's with DNA demethylating agent caused a huge increase in the level of transcripts encoding short interspersed nuclear elements and other species of noncoding RNAs that generated a strong type 1 IFN response. This did not occur in p53 wild-type MEF's. Thus it appears that another function of p53 is to silence repeats that can accidentally induce an immune response.
This has several implications for how we understand self versus non-self discrimination. When pathogen-associated features were quantified, specific repeats in the genome not only display PAMP's capable of stimulating PRRs but, in some instances, have seemingly maintained such features under selection. For organisms with a high degree of epigenetic regulation and chromosomal organization immuno-stimulatory repeats release a danger signal, such as repeats released after p53 mutations. Here, immune stimulation may act as back-up for the failure of other p53 functions such as apoptosis or senescence due to mutation. This supports the hypothesis that specific repeats gained favor by maintaining non-self PAMPs to act as sensors for loss of heterochromatin as an epigenetic checkpoint of quality control that avoids genome instability generally.
When P53 mutates it begins to fail its restraint of viral suppression, this enables a 'viral mimicry' and aberrant immune reactions. These may promote survival of cells that can leverage immunity, promote angiogenesis and heightened proliferation of cancers, or other diseases under modified conditions for non-self tolerance.