|
Immediately prior to fertilization spermatozoa are devoid of Mitochondrial DNA (mtDNA), potentially explaining an aspect about selection that may serve the legacy for maternal immune tolerance. Post fertilization, on day 11-13, outermost trophoblasts of the blastocyst dock with the decidual lining as it embeds in the uterine wall. Then, maternal vascular remodeling and placental formation begin toward successful implantation.
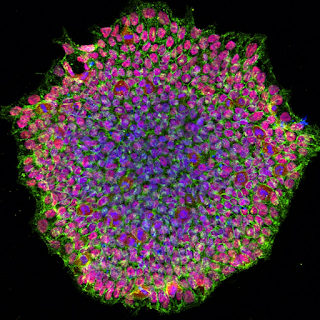
Higher quality trophoblasts are associated with lower mtDNA content. Moreover, euploid blastocysts with higher mtDNA content had a lower chance to implant and mtDNA replication is strictly downregulated between fertilization and the implantation. What is it about absent or reduced mtDNA that may also relate to the mechanics of immune tolerance and vascular remodeling, which are also features of solid tumors.
The initial absence or downregulation of MtDNA, may relate an immune tolerance by uterine Natural Killer (NK) cells. As mtDNA upregulates, after day 12, it may initiate NK auto-reactivity required for maternal microvascular remodeling. This auto-immune paradox is a prerequisite for vascular remodeling, which is also seen in localized hypertension, and the likely basis of successful blastocyst implantation. Acutely, micro-hypertension induced mechanical stretch, on endothelial cells, interconnects innate and adaptive immune responses.
The dominant cell in the decidua is an NK subset (dNK), they express low levels of IFN-γ and express proteins of Renin Angiotensin System (RAS). At day 12 RAS peptide ANP colocalizes to dNK’s suggesting that dNK RAS infers localized responsiveness. When TFAM, required for transcription of mtDNA, was deleted from cardiomyocytes, after 32 days, animals developed cardiomyopathy and Nppa (gene encoding ANP) and Nppb expression was elevated.
In monocytes increased endothelial stretch activates STAT3, which is involved in driving almost all pathways that control NK cytolytic activity and reciprocal regulatory interactions between NK cells and other components of the immune system. The crosstalk between STAT3 and p53/RAS signaling controls cancer cell metastasis. p53, Stat3, and, potentially, the estrogen receptor are thought to act as co-regulators, affecting mitochondrial gene expression through protein-protein interactions. Co-immunoprecipitation of p53 with TFAM suggests it may regulate mitochondrial DNA-damage repair.
Like initial trophoblasts with low level mtDNA, mature cells, like cardiomyocytes that prolong low level mtDNA may also aggravate autoimmune sponsored hypertension that remodels microvascular networks providing nutrients for growth of reduced mtDNA stem cell replicas. Indeed, mitochondrial dysfunction (from depleted mtDNA) does not affect pluripotent gene expression, but results in severe defects in lineage differentiation.
During severe sepsis, intense, on-going mtDNA damage and mitochondrial dysfunction could overwhelm the capacity for mitochondrial biogenesis, leading to a gradual decline in mtDNA levels over time. This may contribute to monocyte immune deactivation, which is associated with adverse clinical outcomes and could be reversed by IFN-γ.
Identifying cells that optimally educate cocultured NK cells for precision IFN-γ and cytolytic responsiveness is part of the ongoing work by the Codondex team.